Debut de sarcoma de Ewing primaro orbitario con parálisis oculomotora del IV par craneal: a propósito de un caso
Dres. Gutiérrez Partida MB1, Franco Iglesias G1, López Tizón E1, Gómez de Liaño Sánchez P1, Merino Sanz P1
1 Instituto Oftálmico. HGH Gregorio Marañón de Madrid.
Introducción
El Sarcoma de Ewing (SE) fue descrito por primera vez por James Ewing en 1921. Es un raro tumor maligno perteneciente al grupo de los tumores neuroectodérmicos primitivos (PNET) que generalmente afecta a la diáfisis de huesos largos y a la pelvis. Raramente puede localizarse extraesqueléticamente afectando a la región paravertebral y meníngea. En la cabeza, las localizaciones más frecuentes son la mandíbula y el maxilar, encontrándose excepcionalmente en la orbita como tumor primario. El pico de edad se produce entre los 5 y 13 años de edad, con un 90% de los casos por debajo de los 30 años. El 64% ocurren en la segunda década. La relación hombre-mujer es 3:2 y existe una predisposición racial, siendo 15 veces más frecuente en los caucásicos que en la raza negra y la asiática. Suelen producirse metástasis en un 75-80% a los dos años del diagnóstico y micro metástasis en un 80%. Clásicamente el tratamiento se realiza con quimioterapia, cirugía y radioterapia.
Descripción del caso
Varón de 36 años que acude a urgencias por presentar diplopía vertical intermitente y progresiva de 4 días de evolución, sin antecedentes personales sistémicos ni oftalmológicos de interés.
La exploración oftalmológica revelaba: AV corregida de 0,9 en AO; MOI: pupilas isocóricas y normorreactivas sin defecto pupila aferente relativo; BMC y FO sin alteración en ambos ojos.
Exploración de la motilidad ocular extrínseca en urgencias:
- Cover test: exotropía de -2º con hipertropía de ojo izquierdo.
- Estudio de diplopía (con luz blanca y cristal rojo): exotropía con hipertropía de OI en todas las posiciones de la mirada.
- Tortícolis cabeza-hombro derecho.
- Bielschowsky hacia el lado izquierdo.
- Ducciones no limitadas.
Se diagnosticó de parálisis oculomotora del IV par del ojo izquierdo. Se realizó interconsulta a Neurología de urgencias y se solicitó TAC órbito-craneal resultando normal. Se remitió a la sección de Estrabismo para seguimiento.
Exploración en la consulta 1 mes después:
- AV cc: 1 difícil AO.
- Estudio de diplopía: exotropía con hipertropia de OI (se compensa con 4 DP base nasal OD y 8 DP base inferior OI), persistiendo el tortícolis cabeza-hombro derecho 10º y Bielschowsky + hacia el lado izquierdo. En la pantalla de Lancaster, sin embargo, se objetivó una paresia del recto inferior del ojo izquierdo (fig. 2) Se realizó tratamiento con toxina botulínica en tres ocasiones (recto inferior de ojo derecho y oblicuo inferior del ojo izquierdo).
A los 9 días de la última inyección el paciente acude a la consulta de estrabismo refiriendo proptosis del ojo izquierdo de 3 días de evolución con dolor ocular (fig. 1). En la exploración se objetivó un exoftalmos con desviación del globo hacia abajo y hacia fuera. La AV, MOI y el fondo de ojo eran normales. Se realiza TAC órbito-craneal urgente (con y sin contraste, figs. 3 y 4) con el diagnóstico de tumoración benigna o de bajo grado de agresividad, tipo hemangioma o linfangioma orbitario sin descartar pseudotumor orbitario o patología linfoproliferativa. Se ingresó al paciente para realización de RMN urgente (fig. 5), cuyo informe concluye con el diagnostico de tumor neurogénico de posible origen benigno, siendo menos probable la existencia de un hemangiopericitoma, pseudotumor o linfoma.
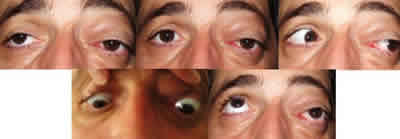
Fig. 1. Imágenes pre-quimioterapia. De superior izquierda a inferior derecha: limitación de la adducción grado -2 OI; exoftalmos con desviación del globo ocular izquierdo hacia abajo y a la izquierda; limitación abducción grado -1; infraversión normal; limitación supraducción grado -2 OI.
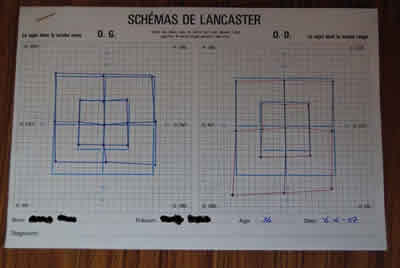
Fig. 2. Pantalla de Lancaster: paresia del recto inferior del ojo izquierdo (diagnóstico diferencial con POM del IV par).
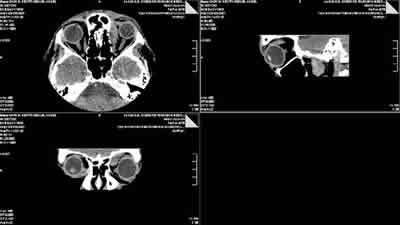
Fig. 3. Primer TAC (sin contraste) al inicio del exoftalmos. Masa extraconal ángulo superointerno de la órbita izquierda.
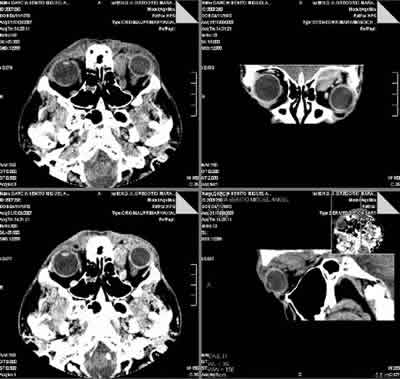
Fig. 4. TAC sin y con contraste.
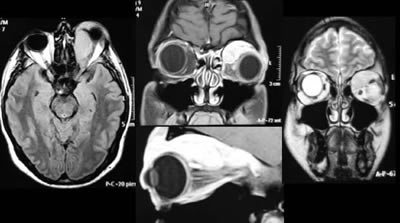
Fig. 5. RMN orbitaria: masa extraconal que se extiende intracranealmente por fisura orbitaria.
Paralelamente a la realización de las pruebas de imagen se exploro al paciente, presentando un empeoramiento del exoftalmos y disminución de la AV cc en tres líneas con el optotipo de Snellen y limitación de todas las ducciones de ojo izquierdo, siendo el resto de la exploración normal. Se instauro tratamiento con 100 mg de prednisona oral al día, sin mejoría de la sintomatología y empeoramiento de AV OI hasta cuenta dedos con FO normal
El servicio de Cirugía Maxilofacial realizó biopsia de la lesión, que se informa por Anatomía Patológica como neoplasia de células redondas de pequeño tamaño, núcleo con grumos finos de cromatina, sin nucleolo marcado y escaso citoplasma vacuolado, fenómenos de apoptosis y marcada actividad mitótica.
Inmunohistoquímica: vicentina+/CD 99 débil+/CD 45-/CD 10-/CD 20-/CD 79 a-/CD 3-/Actina-/E.M.A.-/Citoqueratinas-/Enolasa-/Índice de proliferación celular Ki 67: 80%/Expresión de proteína p53: 30%
Estudio de Citogenético (C.I.N.O.): estudio de citogenética molecular mediante FISH con sonda para el reordenamiento del gen EWS con resultado positivo; diagnostico de Sarcoma de Ewing extraesquelético.
Se estudió por el Comité de Tumores y se realizó estudio de extensión (TAC toracoabdominal y rastreo óseo normal). Se inicio tratamiento con Quimioterapia esquema VAC: vincristina 2 mg/m 2 , adriamicina 75 mg, ciclofosfamida 1.200 mg/m 2 con mesna.
Se realizó nueva exploración oftalmológica, objetivándose amaurosis OI, DPAR OI, úlcera cornal con hipoestesia corneal OI y atrofia óptica con membrana epirretiniana OI.
En la actualidad (fig. 6) el paciente continúa con tratamiento quimioterápico y sigue revisiones oftalmológicas periódicas.
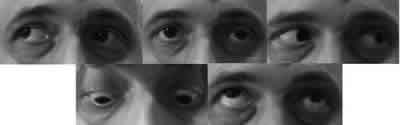
Fig. 6. Imágenes post-quimioterapia.
Discusión
En la literatura existen sólo 20 casos de Sarcoma de Ewing orbitario primario, de los cuales sólo 5 son adultos.
El sarcoma de Ewing es el tumor más indiferenciado de los tumores neuroectodérmicos primitivos y el pronóstico depende principalmente de la existencia o no de metástasis al diagnóstico. Es un tumor característico de los huesos largos y de la edad infantil, pero actualmente existen varias variantes del sarcoma de Ewing.
Los síntomas del sarcoma de Ewing orbitario incluyen exoftalmos, dolor en la región orbitaria, edema palpebral, ptosis y oftalmoplejía y diplopía principalmente. Otros síntomas y signos pueden ser la disminución de la agudeza visual, alteraciones de la motilidad ocular intrínseca, aumento de la presión intraocular y edema de papila. Las manifestaciones más frecuentes son el exoftalmos y la diplopía. Si existe diseminación sistémica pueden presentarse síntomas como la fiebre, la astenia, la pérdida de peso y la anorexia.
Aunque la realización de pruebas de imagen tales como el TAC y la RMN son esenciales a la hora de estudiar una masa obitaria, son insuficientes para realizar el diagnóstico de certeza, realizándose mediante la histología y la inmunohistoquímica, confirmado finalmente por biología molecular. La biopsia puede realizarse mediante PAAF, pero para este tipo de tumores es preferible realizar extirpación de la lesión o parte de la lesión para el estudio anatomopatológico. La histología revela un «sarcoma de células pequeñas redondas y azules». La inmunohistoquímica lo diferencia de otros tumores de células pequeñas, sarcomas y de los linfomas. La biología molecular característica de este tumor consiste en una translocación cromosómica balanceada t(11;22)(q24;q12) que es un marcador fenotípico muy específico. Además existen al menos otras 5 translocaciones en las que el locus 22q12 aparece implicado. Se ha aislado el punto de fractura del cromosoma del Sarcoma de Ewing en el cromosoma 22q12 y se ha aislado el gen EWS (gen del sarcoma de Ewing). Este gen constituye el marcador más importante de este tumor y da el diagnóstico final.
Clásicamente el tratamiento del sarcoma de Ewing se realizaba con cirugía y radioterapia, pero el pronóstico era malo debido a las micrometástasis. Por eso el «éxito» en el tratamiento de este tumor no se consiguió hasta que se trató con quimioterapia. Actualmente, el tratamiento incluye la poliquimioterapia, la cirugía y la radioterapia.
La supervivencia a los 5 años de los pacientes que no tienen metástasis al diagnostico es del 70%; esta cifra desciende al 30% cuando existen metástasis hepáticas y al 20% cuando hay metástasis en otras localizaciones.
Conclusión
Este caso clínico constituye una manifestación atípica del sarcoma de Ewing. Lo que inicialmente se podía confundir con una posible parálisis óculomotora del IV par del ojo izquierdo antigua descompensada en la edad adulta, resultó ser uno de los tumores más indiferenciados que existen.
Todo paciente que debuta con una diplopía brusca debe ser examinado por Oftalmología y debe someterse a pruebas de imagen, siendo el TAC órbito-craneal la primera prueba a realizar. La diplopía puede ser causada por una parálisis oculomotora del III, IV o VI par craneal o por una paresia aislada de uno de los músculos extraoculares. Estos pacientes deben ser evaluados también por Neurología para descartar patología vascular, tumores cerebrales y enfermedades desmielinizantes, entre otras.
Inicialmente, este paciente parecía presentar una parálisis óculomotora del IV par craneal del OI (por la exotropía e hipertropía del OI, el aumento de la diplopía al mirar hacia abajo (bajar escaleras, leer...) con TAC órbitocraneal y estudio oftalmológico y neurológico normal.
Posteriormente, por la evolución y pantalla de Lancaster se observó una paresia del músculo recto inferior del OI. En estos casos, la toxina botulínica constituye el tratamiento inicial, requiriendo algún caso cirugía de estrabismo. La aparición de un exoftalmos brusco días después de la inyección de toxina puede hacer pensar en la existencia de un hematoma orbitario o sangrado de una malformación de muy pequeño tamaño no visible por pruebas de imagen, tal como un hemangioma cavernoso. Descartada esta entidad, hay que realizar el diagnóstico diferencial de una masa orbitaria (pseudotumor orbitario, hemangioma cavernoso, linfoma y rabdomiosarcoma). La realización de una RMN es imprescindible ante la existencia de una tumoración orbitaria así como la realización de biopsia mediante PAAF (a veces la muestra es insuficiente), o por cirugía para el Sarcoma de Ewing.
Ante una tumoración orbitaria de crecimiento rápido hay que tener en cuenta el pseudotumor orbitario, patología benigna inflamatoria que mejora con la administración de megadosis de corticoides (como toda patología tumoral orbitaria que tenga componente inflamatorio) produciendo a su vez una descompresión orbitaria médica. En nuestro caso, no hubo mejoría tras la administración (no requería descompresión ya que no presentaba alteración del nervio óptico ni por oftalmoscopía ni por pruebas de imagen).
La patología de superficie ocular también es frecuente en estos pacientes por el gran exoftalmos y la aparición de alteraciones de la sensibilidad corneal.
Como conclusión podemos establecer que es imprescindible la realización de una prueba de imagen (inicialmente TAC órbito-craneal) en pacientes con diplopía y oftalmoplejía, así como valoración por Neurología y seguimiento por unidad especializada de motilidad ocular. Tras el seguimiento de este caso clínico se puede pensar que una evolución atípica (aunque un TAC órbito-craneal sea normal inicialmente) marcará la necesidad de la realización de una nueva prueba de imagen (ante la sospecha de tumoración, fístula, malformación arteriovenosa, ) que no se veía inicialmente.
Inicialmente, el pronóstico vital a los 5 años de este paciente debe ser del 70%, ya que no presentaba metástasis al inicio.
Bibliografía
- Pang, Noelene K, Bartley, George B. Primary Ewing Sarcoma of tshe Orbit in an adult. Ophthal Plast Reconstr Surg 2007; 23: 153-154.
- Guzowsky, Magdalena B Sc, et al. Primary Orbital Ewing Sarcoma in a Middle-Aged man. Ophthal Plast Reconstr Surg 2005; 21: 449-451.
- Dutton JJ, Rose JG Jr, De Backer CM, Gayre G. Orbital Ewing's Sarcoma of thr Orbit. Ophthal Plast Reconstr Surg 2000; 16: 292-300.
- Wilson D, Dailey R, Griffeth M, Newton C. Primary Ewing Sarcoma of the Orbit. Ophthal Plast Reconstr Surg 2001; 17: 300-303.
- Li T, Goldberg RA, Becker B, McCann J. Primary Orbital Extraskeletal Ewing Sarcoma. Arch Ophthalmol 2003; 121: 1049-1051.
- Lam DSC, Li CK, Cheng LL, et al. Primary orbital Ewing 's Sarcoma:report of a case and review of the literature. Eye 1999; 13: 38 -42.
- Choi RY, Lucarelli MJ, Imesch PD, et al. Primary Orbital Ewing sarcoma in a middle-aged woman. Arch Ophthalmol 1999; 117: 535-537.
- Bajaj MS, Pushker N, Sen S, et al. Primary Ewing s Sarcoma of the orbit:a are presentation. J Pediatr Ophthalmol Strabismus 2003; 40: 101-104.
- Woodruff G, Thorner P, Skarf B. Primary Ewing s Sarcoma of the orbit presenting with visual loss. Br J Ophthalmol 1988; 72: 786-792.
- Turc-Carel C, Philip I, Berger MP, et al. Chromosomal translocations in Ewing 's Sarcoma. N Engl J Med 1983; 309: 497-498.
- Ahmad R, Mayol BR, Davis M, Rougralff BT. Extraskeletal Ewing 's Sarcoma. Cancer 1999; 85: 725-731.
- Chao TK, Cahng YL, Sheen TS. Extraskeletal Ewing 's Sarcoma of the scalp. J Laryngol Otol 2000; 114: 73-75.
- Alvarez-Berdecia A, Schult L, Bruce DA. Localized Primary Intracranial Ewins s Sarcoma of the Orbital roof. J Neurosurg 1979; 50: 811-813.
- Granowetter L, West DC . The Ewing s Sarcoma family of tumors.Ewing s Sarcoma and peripheral primitive neuroectodermal tumor of bone and soft tissue. Cancer Treat Res 1997; 92: 253-308.
- Albert DM, Rubenstein RA, Scheie HG. Tumor metastasis of the eye. Am J Ophthalmol 1967; 63: 727-732.
- Meyers PA, Levy AS. Ewing 's Sarcoma. Curr Treat Opt Oncol 2000; 1: 247-257.