Síndrome de Alport (a propósito de un caso)
Dres. Conesa Hernández E1, Otero Villena MJ1, Martínez Garchitorena J1
1 Hospital Central Cruz Roja. Madrid.
Epidemiología
Incidencia de 1/10.000 habitantes en la población general.
No hay descrita mayor prevalencia en ningún grupo étnico concreto.
Herencia
Defectos genéticos
Forma Clásica: cromosoma X; 85% de los casos mutación del gen COL4A5: Xq21-q22.
Forma autosómicas: recesivas y dominantes; 15% de los casos (AR) alteraciones en los genes COL4A3 y COL4A4 en cromosomas 2 y 13.
Defectos moleculares
Alteración (principalmente) de los genes que codifican las cadenas alfa (2, 4, 5 y 6) del colágeno IV.
El colágeno IV es un componente fundamental de las membranas basales (presente en glomérulo, cóclea, cápsula anterior, cristalino, retina, pulmón...). Su unidad estructural es la triple hélice.
Gen para a1 y a2: cromosoma 13q34.
Gen para a3 y a4: cromosoma 2q35-37.
Gen para a5 y a6: cromosoma Xq22.
HAD se desconoce el gen afecto.
Una cadena a alterada desestabiliza las demás aunque éstas no tengan defecto genético primario: a5 desestabilizaría a3 y a4, con mayor proporción de las cadenas fetales a1 y a2 que inducen actividad proteolítica por diversas captesinas provocando fragmentación de membranas basales.
Entre los defectos genéticos encontrados (>300) hay desde importantes delecciones hasta mutaciones puntiformes.
Variaciones fenotípicas de este síndrome (fig. 1).
Fig. 1.
Diagnóstico
Se requieren tres de cuatro criterios:
- Historia familiar positiva.
- Hallazgos glomerulares característicos.
- Sordera neurosensorial.
- Anormalidades oculares.
Sospecha de SA ante:
- Manchas blancoamarillentas retinianas + AF de IR (compatible con otras nefritis diferentes).
- Lenticono anterior.
Clínica general
Afectación renal: manifestación principal, es la nefritis hereditaria más frecuente. Lo más característico es la hematuria crónica con proteinuria que progresa a insuficiencia renal.
Afectación auditiva: sordera neurosensorial progresiva (>60% de los enfermos), más común en los varones. Afecta sobre todo las frecuencias altas o agudas (4.000-8.000 Hz) por degeneración de la estría vascular y las células ciliadas del órgano de Corti (fig. 2).
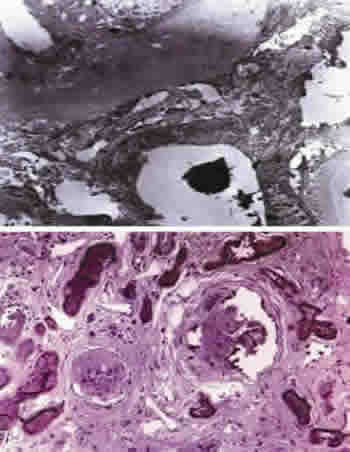
Fig. 2.
Afectación oftalmológica: en 10-30% de los pacientes. Variada y en presencia normalmente de sordera neurosensorial.
Leiomiomatosis difusa: en esófago, tráquea,... En algunos pacientes con formas ligadas al cromosoma X. Se afectan genes COL4A 5 y 6.
Anomalías hematológicas: raras, trombocitopenia.
Variable (de microhematuria aislada a IR terminal).
En principio no específica de una determinada forma genética.
Difícil es predecir la evolución (formas no progresivas y progresivas), en general, el incremento de proteinuria y del defecto auditivo sí se asocian al progreso del fallo renal.
Clínica oftalmológica
Aparece en el 10-30% de los enfermos y su frecuencia se incrementa con la edad.
No hay manifestaciones diferentes entre formas autosómicas y formas ligadas al cromosoma X.
Penetrancia variable.
¿Patognomónico el lenticono anterior (?) si se acompaña de nefritis?
Dianas oculares
Córnea.
Cristalino.
Retina.
Córnea
Vesículas endoteliales compatibles con dismorfia polimorfa posterior (gen TCF8) (fig. 3).
Opacidades subepiteliales (fig. 3).
Arco senil
Microcórnea.
ECR.
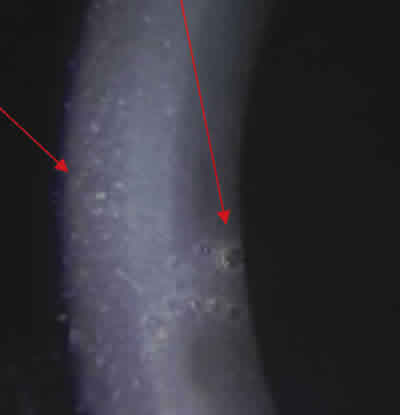
Fig. 3.
Conjuntiva
Calcificaciones subconjuntivales debido a la hipercalcemia por la insuficiencia renal.
Otros signos de polo anterior
Atrofia variable de iris.
Dispersión pigmentaria.
Glaucoma de ángulo cerrado.
Cristalino
- Lenticono anterior bilateral: protusión crónica de la corteza anterior con alteraciones en cadenas a 5,3 y 4.
Constituye un signo tardío.
Afecta al 25% de los enfermos varones adultos.
Se asocia a síndrome de Alport en más del 90% de casos. - Fragilidad capsular (adelgazamiento histológico de la lámina basal y dehiscencia zonular) como base del progreso del lenticono y de catarata polar anterior.
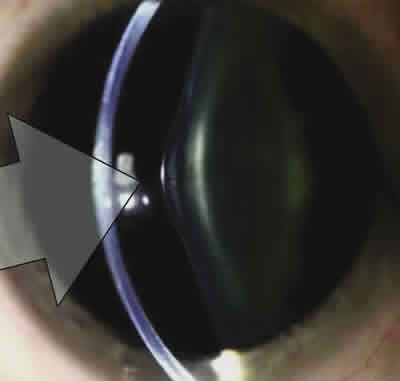
Fig. 4.
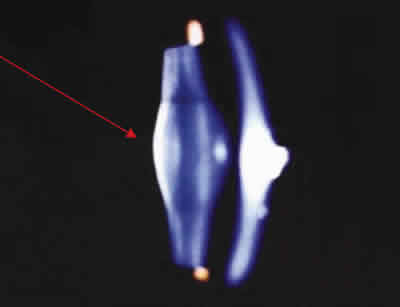
Fig. 5.
Cristalino
Descritos casos de rotura espontánea de cápsula anterior y mayor frecuencia de roturas traumáticas.
Más raros: lenticono posterior (no se asocia a patología sistémica), catarata (a menos que sea cortisónica).
Polo posterior
El 85% de los varones con forma ligada al sexo presentan manchas blanco-amarillentas en polo posterior (no común en la infancia).
Atenuación reflejo macular.
Lesiones maculares similares a distrofia de conos.
Más raro: drusas del NO, retinitis pigmentosa, agujeros maculares.
Caso clínico
Motivo de consulta
Varón de 48 años que es remitido a nuestro centro por baja agudeza visual.
Antecedentes personales
Acude con el diagnóstico de Alport renal y auditivo.
HTA en TTO.
Diálisis peritoneal desde 2003.
Hiperuricemia con crisis gotosas >10 años de evolución.
Hipoacusia bilateral.
Miopía desde la juventud.
VHB+.
Antecedentes familiares
Tres hermanas, una de ellas de 46 años, transplantada de riñón y con antecedente de sordera desde los 20 años. Posibilidad de SA recesivo.
Resto sin interés.
Exploración (fig. 6)
- AV de 0,5 en ambos ojos.
- Refracción: -7,5 -1,75 a 5° (OD) y -9,25 -1,0 a 175° (OI).
- Su graduación (2003): -5,5 -1,25 a 177° (OD) y -7,75 -1,25 a 172° (OI).
- Queratometría: 42,5 D a 0°, 44,25 D a 90° (OD); 42,5 D a 0°, 44D a 90° (OI).
- Eje axial: 24,8 mm (OD), 25,4 mm (OI).
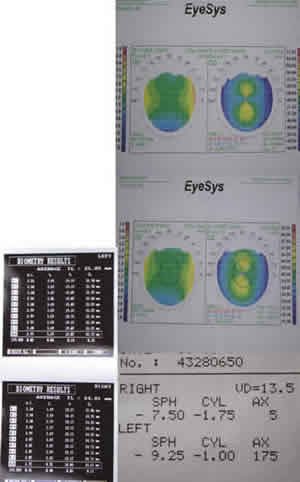
Fig. 6.
Polo anterior (fig. 7):
- Concreciones cálcicas en conjuntiva tarsal.
- Distrofia endotelial corneal.
- Cristalino transparente.
- Lenticono anterior incipiente ambos ojos.
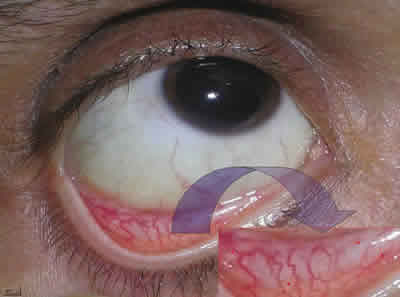
Fig. 7.
Polo posterior
Atenuación reflejo foveal en ambos ojos.
Cambios pigmentarios foveales marcados con aspecto de ojo de buey OD.
Moteado difuso blanco amarillento periférico y central ambos ojos.
Actitud terapéutica
No existe tratamiento específico (terapia génica).
El tratamiento se orienta hacia la clínica desarrollada.
Consejo genético:
- Formas autosómicas: penetrancia del 15%.
- Ligada al cromosoma X: portadores y enfermos (lionización y variabilidad).
Actitud expectante ante este síndrome.
Nueva graduación: mejora de AV a 0,6 dif. en ambos ojos.
Control evolutivo a los 8 meses: se mantiene estable su AV con incremento de 0,25 de miopía.
Seguirá revisiones cada 6 meses.
Se le ofrece la posibilidad de cirugía de cristalino transparente de ambos ojos para evitar su miopización progresiva. Peligro de debilidad capsular con la evolución.
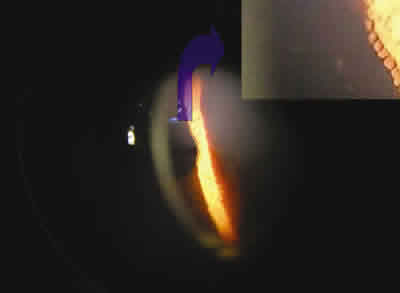
Fig. 8.
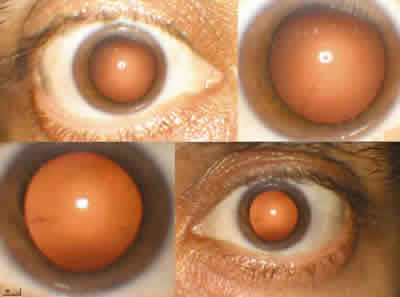
Fig. 9.
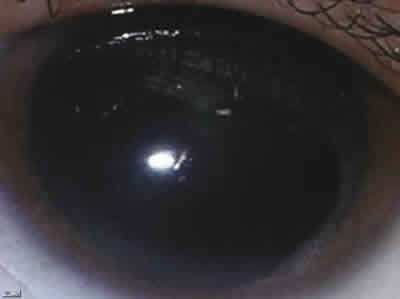
Fig. 10.
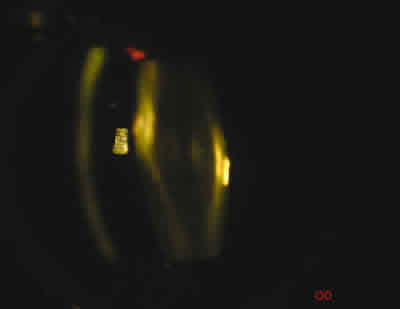
Fig. 11.
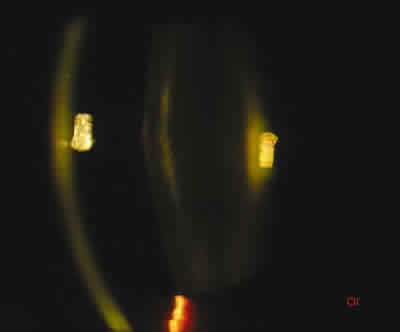
Fig. 12.
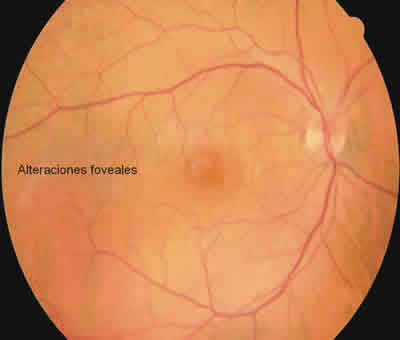
Fig. 13.
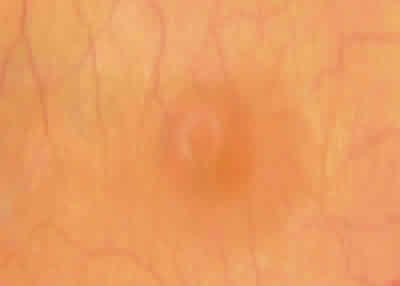
Fig. 14.
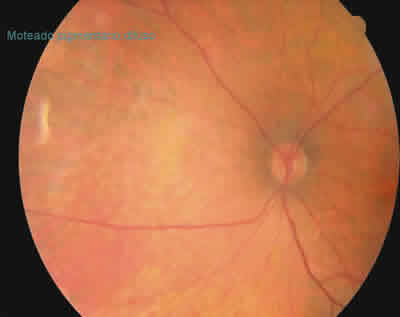
Fig. 15.
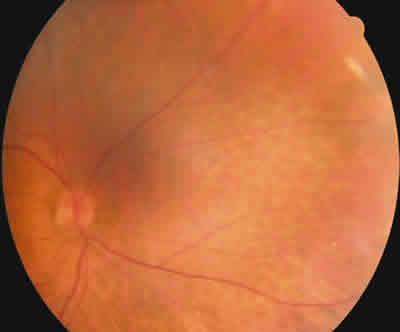
Fig. 16.
Conclusiones
Amplios abanicos genotípico y fenotípico: variabilidad clínica.
Manifestaciones oculares inconstantes y tardías por lo general (mejor pronóstico).
Disminución de AV por miopización progresiva.
Posibilidad de cirugía refractiva.