Hamartoma combinado de epitelio pigmentario retiniano y retina. A propósito de un caso
Dres. Bolívar de Miguel G1, Castejón Cervero MA1
1 Hospital Universitario Príncipe de Asturias. Alcalá de Henares. Madrid.
Introducción
El hamartoma combinado de epitelio pigmentario retiniano y retina es una tumoración poco frecuente, de carácter benigno, que se debe a un crecimiento de varios constituyentes de la retina como el epitelio pigmentario, elementos vasculares y componente glial. Presenta una apariencia clínica variada pudiendo confundirse con patología maligna, como el melanoma coroideo y el retinoblastoma. Presentamos el caso de una paciente que tras una disminución de agudeza visual fue diagnosticada de un posible hamartoma combinado de epitelio pigmentario retiniano y retina.
Caso clínico
Paciente de 27 años de edad que acude por urgencias al notar una disminución de agudeza visual en el ojo derecho, lo que refiere la paciente es que le cuesta más fijar con dicho ojo, sin poder concretar desde cuándo le ocurre, aunque cree que hace unas semanas y lo relaciona con el embarazo que ha tenido y con la lactancia.
Como antecedentes personales la paciente presenta un glaucoma congénito en el ojo izquierdo, del que ha sido intervenida en varias ocasiones (se le han realizado una goniotomía y dos trabeculectomías), además el ojo izquierdo es un ojo microftálmico y que actualmente presenta una hipertropía, la paciente presenta también una miopía magna y un nistagmus congénito motor bilateral.
Como antecedentes familiares destacan otros tres hermanos afectos de glaucoma congénito, padre y abuelo paterno también con glaucoma.
En la exploración que se realiza en la urgencia vemos que la agudeza visual con su corrección (que es 7 dioptrías en el ojo derecho y 9 dioptrías en el izquierdo) es: en el ojo derecho de 0,3+2, que no mejora con el estenopecio y en el ojo izquierdo la agudeza visual es cuenta dedos a medio metro (la agudeza visual previa de la paciente era de 0,4 en el ojo derecho). El defecto pupilar aferente relativo no se puede valorar debido a las sinequias posteriores que presenta el ojo izquierdo y se aprecia una hipertropía izquierda de unos 20º. En la biomicroscopía anterior no se observan alteraciones en el ojo derecho mientras que en el izquierdo se aprecia una microcórnea, cambios cataratosos incipientes y sinequias posteriores, con iridectomías. Las presiones intraoculares eran de 14 milímetros de mercurio en el ojo derecho en tratamiento con timolol y en el ojo izquierdo de 20 milímetros de mercurio. Tras dilatar a la paciente se observa en el fondo de ojo derecho una lesión a nivel de la zona superior de la papila con aspecto sobreelevado, con alguna zona algo más pigmentada y que distorsiona el borde papilar dando un aspecto de tracción, siendo la periferia normal (fig. 1).
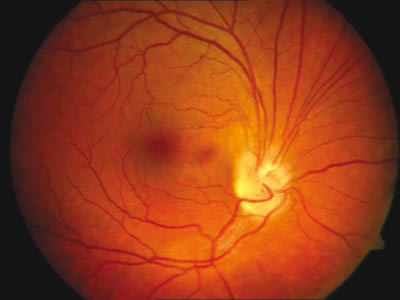
Fig. 1.
Tenemos una imagen previa del fondo de ojo de la paciente, de hace unos quince años, en la que no se observa esta alteración (fig. 2).
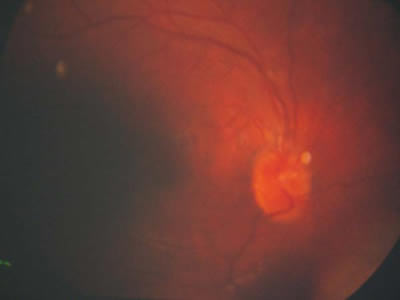
Fig. 2.
Se le realiza a la paciente una angiografía fluoresceína en la que se observa en tiempos precoces alguna zona de hipofluorescencia que coincide con las zonas de mayor pigmentación y una zona de mayor vascularización en la zona superior de la papila (figs. 3 y 4), observándose en tiempos tardíos alguna zona de hiperfluorescencia (fig. 5).
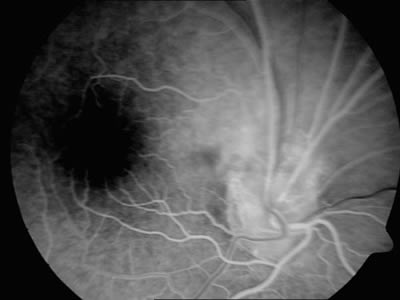
Fig. 3.
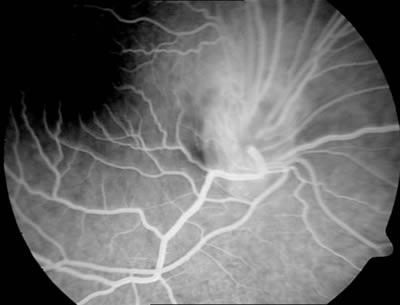
Fig. 4.
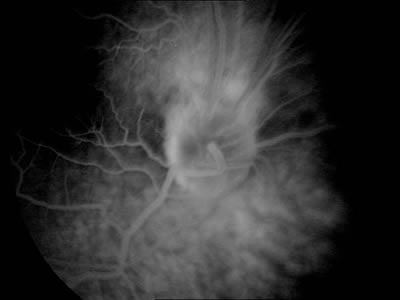
Fig. 5.
Se realiza también un campo visual que teniendo en cuenta que la paciente es una miope magna y con nistagmus no presenta grandes alteraciones y en el que no se aprecian cambios respecto a campos visuales previos.
A la paciente se le realizó una tomografía axial computerizada y una resonancia magnético nuclear que no presentaron alteraciones, sigue en seguimiento sin haber presentado cambios en la agudeza visual ni en la tumoración.
Discusión
El hamartoma combinado de epitelio pigmentario retiniano y retina es una lesión rara aunque benigna.
El primer caso fue descrito por Roveda en 1952 aunque el nombre con el que se conoce actualmente a estas lesiones se le atribuye a Gass en el 1973 (1).
Se cree que son lesiones congénitas o del desarrollo, aunque nunca se han encontrado en recién nacidos y hay alguna lesión que se ha descrito en pacientes con fondo de ojo previamente normales (2). También han sido publicados casos de hamartomas adquiridos, normalmente en paciente que han presentado procesos inflamatorios (3,4).
La prevalencia con que se encuentran estas lesiones es similar en hombres y mujeres, o algo mayor en los hombres, y con un amplio rango de edades de diagnóstico, con una media de 15 ó 18 años según las distintas publicaciones (5-7).
La causa más frecuente por la que consulta el paciente es por una disminución de agudeza visual indolora (que en un 60% de las ocasiones es el motivo de consulta), aunque otras formas de presentación son estrabismo en un 18%, miodesopsias en un 5%, leucocoria y dolor ocular en un 3% de las ocasiones respectivamente, siendo un hallazgo casual durante una revisión rutinaria en el 10% de las ocasiones (5,7).
La agudeza visual varía según la localización de la lesión, la afectación directa del nervio óptico, el haz papilomacular y la fovea disminuyen la agudeza visual; también la disminuyen las membranas epirretinianas que distorsionan la macular. En casi el 45% de los pacientes con hamartoma combinado la agudeza visual es mayor o igual a 20/40 (5).
El diagnóstico que se hace es fundamentalmente clínico, observándose una lesión normalmente unilateral, de localización más frecuente en la zona adyacente al disco óptico, aunque también se pueden encontrar a nivel macular o en la periferia, y que presenta tortuosidad vascular, hiperpigmentación (se han encontrado casos poco o nada pigmentados), con ligera elevación y con formación de membranas epirretinianas (aunque no se encuentran en todos los casos) y en algunos exudación (5-8).
La angiografía puede ayudar bastante en el diagnóstico observándose en las fases precoces hipofluorescencia en relación al grado de hiperpigmentación del tumor, tortuosidad vascular y telangiectasias. En la fase media se observan mejor las anomalías vasculares y según avanzan los tiempos los vasos suelen fugar dando lugar a una hiperfluorescencia tardía (7).
Se han visto múltiples asociaciones oculares (5-7) como son la existencia de un defecto pupilar aferente relativo, estrabismo, hemorragia vítrea, neovascularización coroidea, líquido subretiniano, retinosquisis juvenil ligada al X, fosetas congénitas, colobomas y drusas del nervio óptico, en cuanto a asociaciones a nivel sistémico cabe destacar la neurofibromatosis tanto tipo 1 como 2 [encontrándose la mayoría de los casos de hamartomas bilaterales en pacientes con neurofibromatosis (6,9,10)], hemangioma facial, incontinencia pigmenti, esclerosis tuberosa, síndrome de Gorlin (11), angiofibroma nasofaríngeo juvenil (12), membrana epirretiniana con agujero macular (13).
En cuanto a la evolución clínica es importante tener en cuenta que en algunos de los casos que hay descritos se ha observado un crecimiento de la lesión que llevó a sospechar de una lesión maligna y realizándose por este motivo la enucleación, confirmándose el diagnóstico de hamartoma combinado de epitelio pigmentario retiniano y retina al realizar el estudio histopatológico que es el que nos conduce al diagnóstico definitivo y en el que podemos observar engrosamiento retiniano y del nervio óptico, perdida de la arquitectura normal, y alteraciones a nivel vascular, del epitelio pigmentario retiniano y proliferación glial (1,14).
La agudeza visual disminuye en algunos casos y esto se puede deber a hemorragias, a las membranas epirretinianas..., como tratamiento hay que evitar la ambliopía cuando nos encontramos en edades en que esto es posible, tratar las hemorragias vítreas cuando ocurran y también se han realizado tratamientos de las membranas epirretinianas con resultados variables, aunque en la mayoría de los casos la agudeza visual no mejora mucho (5,7,14).
Bibliografía
- Gass JDM. An unusual hamartoma of the pigment epithelium and retina simulating choroidal melanoma and retinoblastoma. Ophthalmology 1984; 1: 1609-1619.
- McLean EB. Hamartoma of the retinal pigment epithelium. American Journal of Ophthalmology. 1976; 82: 227-231.
- Ticho BH, Egel RT, Jampol LM. Journal of Pediatric Ophthalmology and Strabismus. 1998; 35: 116-118.
- Hrisomalos NF, Mansour AM, Jampol LM, Fowell SM, Greenwald MJ. «Pseudo» -combined hamartoma following papilledema. Archives of Ophthalmology 1987; 105: 1634-1635.
- Schachat AP, Shields JA, Fine SL, Sanborn GE, Weingeist TA, Valenzuela RE, Brucker AJ. Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology 1984; 91: 1609-1615.
- Font RL, Moura RA, Shetlar DJ, Martinez JA, McPherson AR. Combined hamartoma of sensory retina and retinal pigment epithelium. Retina 1989; 9: 302-311.
- Ryan. Retina, ed 3, 2001, Mosby.
- Hayasaka S, Kanamori M, Furuse N, Ugomori S, Setogawa T. Combined hamartoma of the retina and retinal pigment epithelium of the juxtapapillary retina and optic disc. Ophthalmologica 1988; 197: 42-46.
- Destro M, D'Amico DJ, Gragoudas ES, Brockhurst RJ, Pinnolis MK, Albert DM, Topping TM, Puliafito CA. Retinal manifestations of neurofibromatosis: diagnosis and management. Archives of Ophthalmology 1991; 109: 662-666.
- Gass JDM. Stereoscopic atlas of maculas diseases: diagnosis and treatmente. ed 3, St Louis, 1987, Mosby.
- Fonseca RA, Dantas MA, Kaga T, Spaide RF. Combined hamartoma of the retina and retinal pigment epithelium associated with juvenil nasopharyngeal angiofibroma. American Journal Ophthalmology 2001; 132: 131-132.
- De Potter P, Stanescu D, Caspers-Velu L, Hofmans A. Combined hamartoma of the retina and retinal pigment peithelium in Gorlin syndrome. Archives of Ophthalmology 2000; 118: 1004-1005.
- Mason JO, Kleiner R. Combined hamartoma of the retina and retinal pigment epithelium associated with epiretinal membrane and macular hole. Retina 1997; 17: 160-162.
- Sappenfield DL, Gitter KA. Surgical intervention for combined retinal-retinal pigment epithelial hamartoma. Retina 1990; 10: 119-124.